
 Intercept Pharmaceuticals (NASDAQ:ICPT) Gilead Sciences, Inc (NASDAQ:GILD) Clovis Oncology, Inc. (NASDAQ:CLVS) Chiasma, Inc. (NASDAQ:CHMA)")
This Market Exclusive report covers all major biotech news from last week as well as any major news expected this week.
Companies covered:
Intercept Pharmaceuticals (NASDAQ:ICPT)
Gilead Sciences, Inc (NASDAQ:GILD)
Clovis Oncology, Inc. (NASDAQ:CLVS)
Wednesday March 30th 2016 – Friday April 8, 2016
The first week of April 2016 in biotechnology saw three significant developments, two of which present buying opportunities. We’ll begin just two days before that, in the dead of night at 3:00am on March 30th.
Opko Health Inc

On March 30th, Opko Health Inc (NYSE:OPK) issued a press release stating that the FDA did not approve its flagship drug candidate, Rayaldee. Instead, it issued a Complete Response Letter, or CRL, citing problems at Opko’s contract manufacturer, Catalent Inc. (NASDAQ:CTLT)
This was a shocker to almost everyone at the time – everyone except the people at Catalent, who for obvious reasons could not disclose what was to come. On March 25th, four days before the expected PDUFA and FDA approval for Rayaldee, Catalent was hit with a Form 483, which is basically a piece of paper that details deficiencies in protocol and/or procedure at a contract manufacturing plant. Neither company has disclosed exactly what the deficiencies were, but we do know that Catalent will be addressing them by April 15th, after which Opko is hoping for an FDA response within 30 days.
If that timeline is basically correct, the next catalyst for Opko should be somewhere between mid May and mid July, giving the FDA a reasonable buffer zone within which to respond. They are notoriously slow after all, so those interested in buying Opko should not be tied to any month-specific time frame for this issue to be cleared up.
What we do know is that the CRL did not mention Rayaldee specifically or the data associated with it. The data on Rayaldee are as stellar as they can be, with no significant side effects. The lack of mention of any issues with data make it clear that the FDA will almost certainly approve Rayaldee once the issues with Catalent are dealt with. The only question is time.
The Science
Before we get into the data itself, first some info on Rayaldee. The scientific name for this drug is modified-release calcifediol (MRC). It is designed to treat a condition called secondary hyperparathyroidism (SHPT). SHPT is itself caused by vitamin D insufficiency, which in turn is caused by chronic kidney disease (CKD). What happens here is that people with failing kidneys can no longer convert vitamin D to its active form – a function normally performed by the kidneys. This leads to vitamin D insufficiency, which causes the parathyroid glands to overproduce a different hormone calling for the production of more vitamin D to its active form. This, unfortunately, the patient’s failing kidneys cannot do, so it leads to a vicious cycle of too much parathyroid hormone in the blood, which leaches calcium out of bones, leading to many other problems.
The solutions available today for this condition are quite poor. They consist mainly of vitamin D supplementation. The problem though is that simply giving the supplement to a patient is like feeding a 10 course gourmet meal to a starving man after weeks of eating nothing. Not enough vitamin D and the disease will continue. Too much leads to toxicity with even worse problems.
Rayaldee solves the problem through modified release, which gives the patient a steady and slow supply of the missing prohormone needed to convert vitamin D to its active form.
The Data
The fact that there have been no serious adverse events related to vitamin D toxicity in CKD patients on Rayaldee speaks volumes. In its last Phase III data presented on April 1, 2016 at the Endo2016 conference, Opko revealed that 95% of patients treated with Rayaldee achieved vitamin D sufficiency, and that Rayaldee could even address stage 4 renal disease at higher doses which were also safe and tolerable for patients. This was not previously known, and adds even more value to Opko’s flagship pipeline candidate.
The situation with Opko now is unique in that data could hardly be any better, and approval is as close to certain as one can get in biotech. The only question is timing, but approval looks likely by July.
The Market
There are currently 8 million people with stage 3 and 4 CKD and at or approaching SHPT due to vitamin D insufficiency in the United States. Estimates are that peak sales for Rayaldee could be around $500 to $600 million annually within 10 to 15 years, and Opko has a patent on Rayaldee until 2028. This is just the US market, so sales could be substantially higher than that, approaching blockbuster thresholds 10 to 15 years out.
With many biotechs there is also a risk of failing to penetrate the market, whether due to competition, inexperienced sales staff, or market rejection of the drug for lack of interest. None of these will likely affect Rayaldee’s eventual launch because, first of all, Opko is not your typical startup biotech filled with industry novices unfamiliar with the territory and lacking connections. Opko is headed by Dr. Phillip Frost, the former CEO of the largest generics company in the world, Teva Pharmaceuticals Industries, Ltd. (NYSE:TEVA) Frost and his lieutenants know the marketing landscape well.
Frost catapulted his career by taking over a failing company called Key Pharmaceuticals, successfully remarketing its failing asthma drug way back in the early 1970’s, and from there went on his path to becoming the CEO of Big Pharma’s largest generics giant.
Second, Rayaldee will have no competition. There currently is no effective long term treatment for SHPT associated with CKD. While Vitamin D supplementation is currently used with limited success, it is really no match for Rayaldee’s data due to the toxicity problems associated with standard vitamin D supplementation. The drug is taken orally in gel caps, so patient compliance will not be a major issue either.
Sales of Rayaldee won’t immediately reach $500 million annually, but they will most likely get there within a decade.
The Market Reaction
Opko shares reacted to the disappointing news quite well, considering it was unexpected. The share price was quite resilient. The stock is essentially right back to where it was before the disappointing CRL was issued, and that was less than two weeks ago. If shares can be essentially unaffected by an unexpected delay in approval, that points towards approval not yet being priced in to the shares. If it were, then we would have seen a strong and sudden drop in Opko’s stock, which would have been sustained until the market became aware of a new date for potential approval.
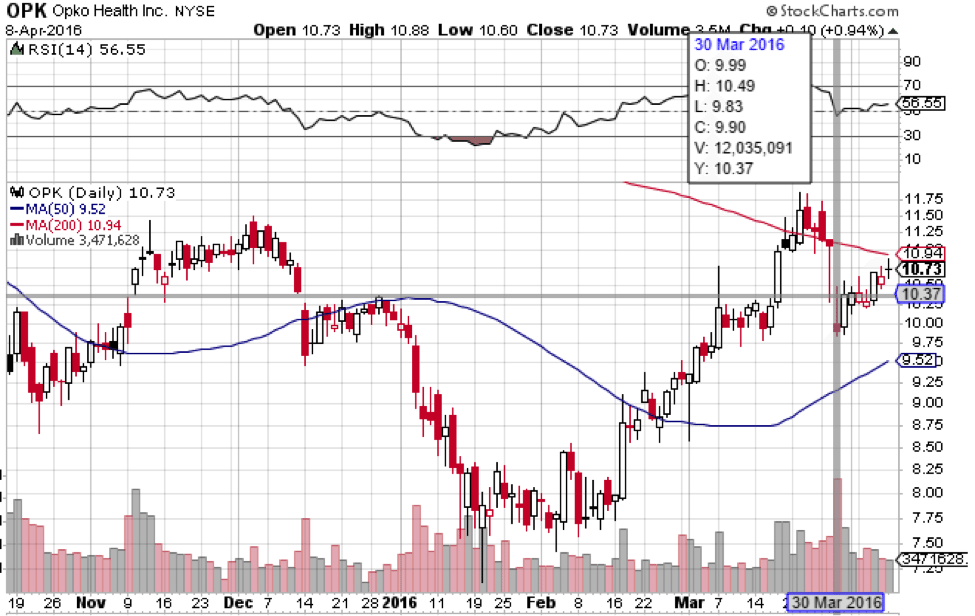
That is not what happened. On the contrary, we saw a relatively small drop of 12% counting from March 29th highs to March 30th lows. That may seem like quite a bit, but consider this: Counting from March 29th lows before news came out to March 30th highs afterwards, we actually saw a rise from $10.28 to $10.49 a share. From that perspective, the market was basically unaffected by the news. Shares are currently at $10.84.
Finances
As the current pet project of billionaire Dr. Frost and multi millionaire Dr. Jane Hsaio who together own over 200 million shares, Opko does not have any immediate cash issues. It currently has about $390 million in cash and net receivables, and thanks to its recent acquisition of Bio-Reference Laboratories, is net positive for now. Its continuing to be net positive depends on Rayaldee’s eventual approval this year and a successful sales launch of course, as despite being net profitable, it still has a negative operating income.
Target and Trading Strategy
Opko tends to move together with the wider biotechnology index. If you look at a chart of Opko versus the iShares NASDAQ Biotechnology Index ETF (NASDAQ:IBB), you’ll see a strong correlation. Both Opko and the IBB topped at around the same time, meaning that the market treats Opko as more than just a development trial-stage company dependent on its own data. With momentum in biotech reversing from its bearish trend since July 2015, Opko can easily head back up near its old highs at $19.20, which were hit without any FDA approval.
Once Rayaldee gets the go-ahead, a near term target of $20.00 a share is reasonable, which would put market cap at between $11 and $12 billion. Such a target would be easy to reach from a technical perspective considering the stock’s past, but fundamentally it would be a little high considering that Rayaldee does not quite have blockbuster potential. Even assuming Rayaldee hits $500 million in sales annually next year, which it won’t, total revenues for the company would be around $800 million.
A market cap 10 times revenues is a high number, so a reasonable strategy would be to go overweight now, but then to take profits and lighten positions as Opko approaches its all time highs. However, zeroing positions at $20 is not recommended, as Opko has a diversified portfolio with much more potential than just Rayaldee, including a long-acting human growth hormone candidate in Phase III, scheduled for primary completion in August.
Intercept Pharmaceuticals
![]()
On April 7th, an FDA panel unanimously recommended approval for Ocaliva, a treatment for primary biliary cirrhosis (PBC) developed by Intercept Pharmaceuticals Inc. (NASDAQ:ICPT).
The Science
PBC is a lethal liver disease that results from autoimmune destruction of the bile duct. The bile duct connects the liver to the intestine, transporting bile acids (BAs) out of the liver and into the intestine to aid digestion. When the immune system attacks the bile duct, BAs leak out of the bile duct into the blood, and they also back up into the liver. This causes toxic build-up of BAs into the liver, eventually leading to cirrhosis and liver failure. Disease progression is relatively slow, but eventually requires liver transplantation within 5 to 10 years.
Ocaliva’s scientific name is obeticholic acid (OCA), and it is a farnesoid x receptor (FXR). The FXR is a receptor that is naturally activated by BAs in the blood, which in turn shuts down further production of bile by the liver. OCA is a more powerful FXR that shuts down bile production faster and more efficiently than bile itself. Note that OCA does not actually repair the damage that the immune system inflicts on the bile duct. Rather, it only serves to decrease bile acid production by the liver so the liver doesn’t get damaged by bile acid backup.
The Data
The story with OCA and Intercept goes back to January 9, 2014, when a Phase 2b trial for the drug was halted prematurely because it had already met its primary endpoint after an interim analysis. That trial was a little bit different because it was for a slightly different liver disease indication. The trial back in 2014 was for non alcoholic steatohepatitis, which is basically the same thing as cirrhosis from alcoholism but instead of being caused by excessive consumption of alcohol, it’s due to excessive consumption of fat and sugar.
OCA as an FXR also speeds up the metabolism of fat in the liver, which is why it seems to help the condition. However, the problem is that since OCA slows down bile acid production by preventing cholesterol-based compounds from being turned into bile, it also raises blood cholesterol, a serious problem for patients who already have trouble with high fat and high sugar diets. This was also one of the reasons that the trial was suspended – both because it met its primary endpoint, and also because the side effects were actually somewhat dangerous, a rare combination in such a successful trial indeed.
Intercept’s press release on the matter predictably only emphasized the good part, namely that the trial was stopped because it had already reached its endpoint. The company chose not to emphasize the problematic portion of the trial.
As for the data on the PBC indication, the an FDA panel unanimously recommended approval on last week, it is less problematic, though still slightly so. 46% of patients on the lower 5mg dose met the primary endpoint, being lower alkaline phosphatase (ALP) in the blood, a biomarker of liver disease. 47% met the same endpoint on the 10mg dose. While that makes it look like it doesn’t work for everyone, still 98% of patients either improved or stabilized ALP.
The Market
1 in 1000 women over 40 are estimated to have PBC. The total US market is estimated at 174,000, with some analysts seeing a price point of between $11 and $20,000 a year for patients. At 100% market penetration, that translates to between $2 and $3.5 billion annually in revenues, but of course Intercept will not reach 100% market penetration. There are still cholesterol problems associated with OCA, and though side effects were not serious in the PBC indication, pruritis, or skin itching, is still a common side effect with this medication. This means that some patients may have a hard time taking it and may prefer not to.
Still, on the plus side Intercept will have little competition in the market as it will be only the second approved drug for PBC, and a unanimous board recommendation for approval all but assures that will happen. The question will be what happens when the drug gets to market and how much will it sell on a consistent basis.
The only other approved drug on the market for PBC is ursodiol, which is also known to improve liver chemistry as far back as 1991. Like OCA, ursodiol improves ALP levels and bilirubin levels in the blood, which also indicates disease reduction. However, ursodiol has never been shown to improve clinical outcomes significantly. That said, OCA does seem to be more effective than ursodiol in lowering disease markers for a longer period of time. Specifically, 35% of patients in the placebo/ursodiol arm experienced increases in ALP after 12 months of treatment, while levels stayed down in the OCA/ursodiol arm. Nevertheless, long term studies have not been completed in order to determine if OCA is indeed better than ursodiol in improving clinical outcomes.
Such a trial is underway, nicknamed the COBALT trial, following patients on OCA for up to 8 years. Intercept does not have to succeed with this trial to get approval, but results will speak volumes for sales of the drug, and we are a long way from knowing definitively if OCA actually improves disease outcomes over a long period of time.
The Market Reaction
Back in 2014, the market did indeed overreact to OCA’s Phase 2b success with NASH liver disease (see chart below). Shares rocketed 620% overnight as everyone focused on the fact that OCA met its primary endpoint but ignored the implication of a rise in serum cholesterol. Shares flew into the stratosphere way out of proportion to highs of $497 a share and have been falling ever since.
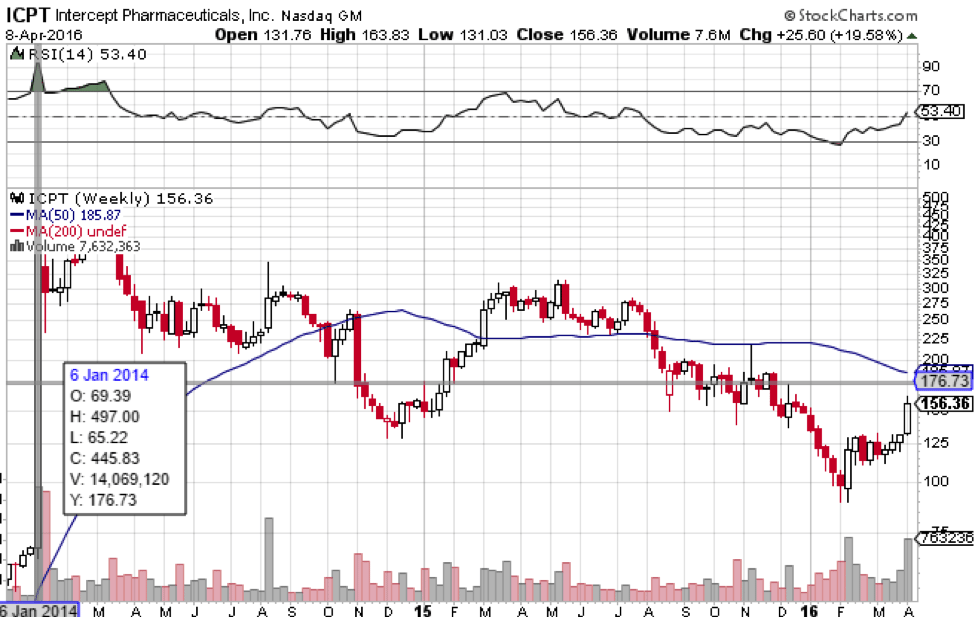
That is a big reason why market reaction to the news of unanimous recommendation for approval of OCA for PBC was muted. Trading on April 7th was halted and shares opened higher the next day, but fell back down the same day. The market was not going to be fooled again by an overreaction to the same drug. We’d say the lack of enthusiasm for what is otherwise good news is indicative of market suspicion that this may not spell success for Intercept financially in the end. While it might, the uncertainty surrounding OCA is palpable, as the hangover from January 2014 when shares went to $497 is still in effect. As of this morning, April 11th, shares are trading for just below $157.
Finances
Money is not an especially pressing issue for Intercept at the moment. That’s the good news. With $642 million in cash and short term investments, the company is well capitalized with an average quarterly burn rate of $56 million over the last year. That means there is plenty of time for OCA to test the market waters before Intercept needs to refinance. The risk is not cash. It’s the market. If OCA fails to sell well in its first year or two, the company will be facing an embarrassing situation of having to dilute its float in order to continue pushing the same candidate that will have already done poorly in the PBC market. By then, investors may have already lost faith in Intercept’s prospects.
The future of Intercept depends on market acceptance of OCA for PBC, not FDA approval. Market acceptance may indeed happen and all may be well, but the point is that FDA approval is not really a significant catalyst for the stock. This is already assumed. Patients may not like the prospect of scratching themselves due to pruritis when they cannot even be assured definitively that the drug will help prolong their lives or improve their quality of life.
Valuation and Trading Strategy
Intercept is currently valued at $3.8 billion. Even assuming a valuation of 10 times revenues let alone earnings – again quite a high figure – OCA will have to sell about $380 million a year to justify Intercept’s valuation as it currently is. That translates to a market penetration of about 10% at the upper price point of $20,000 annually. At the low end, It translates to close to 20% market penetration, and that is at the valuation we are already at pre-approval.
The most we can say about Intercept at this point is that it is a speculative position with limited upside potential from here. A small underweighted position can be held in case OCA succeeds in the market and ultimately proves clinical outcomes, which would pave the way for the success of OCA in the NASH indication, and that is indeed blockbuster. The prospects of this happening, at least as it stands now, appear slim, though not infinitesimally so. Once again though, if you are trading on catalysts, it doesn’t appear that FDA approval for OCA will spur a sustained rally in Intercept shares.
Gilead Sciences

On April 4th, the FDA approved Descovy, an antiretroviral combination drug for the treatment of HIV, developed by Gilead Sciences, Inc. (NASDAQ:GILD). Descovy’s scientific name is F/TAF for Emtricitabine/Tenofovir Alafenamide. This follows positive Phase III results announced by Gilead back in September.
The Science
F/TAF is a reverse transcriptase inhibitor, an enzyme that HIV uses to replicate itself once its DNA invades a host cell. What is special about F/TAF is that unlike some other reverse transcriptase inhibitors, it is a prodrug that only becomes active once inside a cell, and it is more efficient at getting into cells at relatively low concentrations in the blood. In fact, studies have shown that F/TAF is as effective as its predecessor TDF at serum concentrations up to 90% lower.
Unfortunately, F/TAF is still not a cure for HIV. That has not yet been discovered because HIV’s replication process ironically produces transcription errors in its DNA/RNA, which leads to fast mutation of the disease and the resulting inability to target a specific genome with a stable vaccine. Still, F/TAF can now serve as the backbone for drug cocktail regimens for the HIV population at considerably less risk than currently used antiretroviral drugs.
The Data
The Phase III studies off of which Descovy was approved showed statistical noninferiority to TDF, the currently most popular antiretroviral backbone drug. One Phase III study focused on patients that have had no prior treatment of their HIV infection. The second focused on patients that were already virologically suppressed and switched from TDF to TAF. Results showed continued suppression of less than 50 copies of the virus per mL of blood, but with improved kidney function and bone density. Since drug concentrations are so much lower than TDF regimens, the kidneys do not have to clear as much of it from the blood, so they are less stressed than they are with TDF.
The trial testing patients already virologically suppressed enrolled 665 patients over a treatment period of 96 weeks. The treatment-naïve trial tested 600 adults for the same time period, almost 2 years.
The Market
Before we get to specific numbers, it is important to realize that Descovy’s success as an F/TAF is not new. Gilead already has an F/TAF drug on the market called Genvoya. As successful as Genvoya was in the clinic, it is still new and has not yet caught on in the market. Genvoya’s cost is $3,200 a month, and Descovy’s pricing will be similar.
HIV is an $18 billion market annually and Gilead dominates that market with $11 billion in sales of its HIV products a year. Genvoya, however, is a tiny percentage of that total at only $45 million in sales last year. This doesn’t mean that Genvoya and other F/TAF’s like Descovy won’t soon be popular or even blockbuster. Rather, it is mostly because Genvoya is brand new, having only been approved in November, so it only had one month to accumulate 2015 sales figures.
Still, Descovy’s sales will probably not be a home run initially because it will be competing with Gilead’s other already popular HIV product line, as well as with Genvoya itself. With Descovy though, the exact sales figures in the first few years are less important, because however the market responds, whether it hangs on to TDF more strongly or Descovy sales are lost to Genvoya, the revenues go to Gilead anyway.
What is important is that with a new generation of antiretrovirals of which Descovy and Genvoya are part, Gilead has cemented its position as the undisputed leader in the HIV space. It is always better to have two products in a given line instead of one, so that patients who may be sensitive to one for whatever reason have the option of taking the other instead. Over time though, F/TAF drugs like Genvoya and Descovy should begin to overtake TDF sales because they are obviously superior given the lower dosage and side effect profile.
Another advantage, also important for Gilead, is that F/TAF drugs can be taken in combination with hepatitis C drugs, which are Gilead’s main product line. This is not the case with TDF drugs.
The Market Reaction
As expected, Gilead shares did not react to Descovy’s approval in any noticeable way. After all, Gilead already has an F/TAF on the market, Genvoya has not taken off yet, and any revenues will mostly be cannibalized from its other HIV products anyway. However, the approval is still a big buying opportunity because it puts Gilead in a continued strong position from which it will not likely be dislodged any time soon.
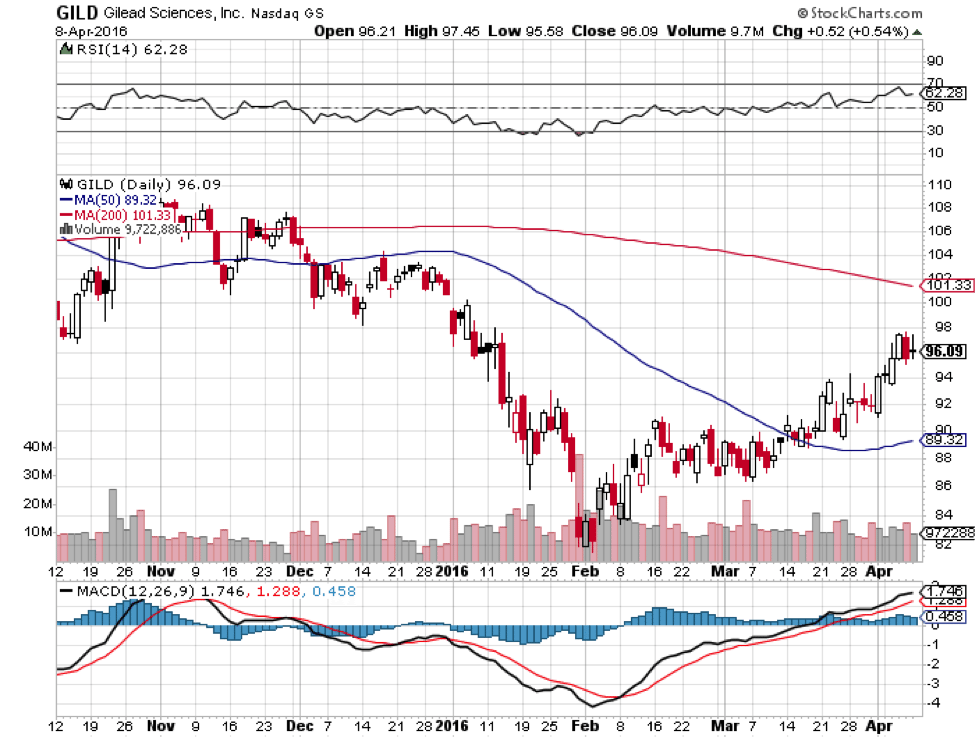
The big danger for Gilead prior to F/TAF was that GlaxoSmithKline PLC (NYSE:GSK) and Pfizer Inc. (NYSE:PFE) would overtake Gilead with their own HIV drug Triumeq, which is already blockbuster at £730 million, (see page 17) or $1.03 billion, in only its second year of sales. Triumeq is statistically superior to Atripla, Gilead’s second bestselling HIV TDF drug behind Truvada. Sales of Atripla have declined 14% since 2013. However, Triumeq does lead to serious side effects in more that 1% of patients, a significant number. With Descovy and Genvoya, Gilead can now stand up to Glaxo and Pfizer’s encroachment on its market.
This makes Descovy’s approval very significant, more than the market is currently letting on. The results are more long term, but should not be underestimated.
Finances
Money is not an issue with an established player like Gilead. The only thing of note, and it is a positive, is that Gilead’s debt load of $22 billion is protected from rising interest rates because it is all fixed-rate debt. At a market cap of $130 billion, debt is insignificant.
Valuation and Trading Strategy
Gilead is 28% below its 52-week high and generally follows the broader biotech sector. It is a good buy at these prices considering its second-largest market is secured from threats thanks in part to Descovy’s approval. Those interested in taking a position should definitely buy sooner rather than later, as biotech seems to have bottomed along with the rest of the broader stock market. Conservative investors can ride Gilead 20% higher to a target of $115 a share with a proportionally weighted position as biotech continues to recover from its multi-month correction. More risk-oriented investors can take a long term multi-year position to new highs if they can commit to riding out the volatility inherent in this sector.
The Week Ahead
Monday April 11, 2016 – Friday April 15, 2016
Here’s a look at what we’re keeping an eye on this week in the development stage biotech space.
04/12/2016
Clovis Oncology, Inc. (NASDAQ:CLVS)
![]()
FDA panel to review Rociletinib for mutant EGFR non-small cell lung cancer
On April 12, 2015, an independent advisory panel is set to review, and vote upon, Rociletinib. The drug is Clovis’ non-small cell lung cancer (NSCLC) candidate, and as many reading will know, it’s had a pretty checkered development path.
When Clovis first announced the results of the phase III on which the NDA is based, the company reported what looked like great efficacy data. However, shortly after, the FDA announced it was going to base its decision on confirmed responses only, as opposed to confirmed and unconfirmed, and that it required an extension on the data provided in the first submission. Concurrent to the FDA announcement, Clovis reported that the confirmed response data was as much as 30% weaker than it had first reported, and the company’s market capitalization took a real hit as a result. There has since been some suggestion that Clovis knew about the disparity between the confirmed data and the unconfirmed data weeks before announcing it, and this suggestion has weighed heavily on market sentiment towards the company.
But that’s not all. The lower confirmed response rate puts the drug at a lower efficacy than a competing drug – AstraZeneca plc’s (ADR)(NYSE:AZN) Tagrisso – which the FDA approved a few days before the Rociletinib debacle hit the press.
The question now, is whether an advisory panel will see any justification for recommending approval of a drug that already has an approved therapy of the same type. Both Tagrisso and Rociletinib are EGFR tyrosine kinase inhibitors, and based on the revised response rates, less effective.
The answer, to be noncommittal, is maybe. Over the last decade or so, the FDA has taken a pretty lenient stance on oncology drugs that have limited treatment options. With Rociletinib having a favorable safety profile, the FDA might give the drug the green light just to flesh out the potential pool of available therapies.
What impact will an approval recommendation have on Clovis’ market capitalization, and conversely, what will the opposite decision mean for the company?
Analysts put the NSCLC market that Clovis is targeting at around $3 billion, and a recommendation would likely lead to a favorable FDA decision. The chance to compete with Tagrissa for a $3 billion market will probably inject some upside momentum into Clovis, and a favorable AP review will likely close the gap on the post-November lost value. With this in mind, a positive review could boost the company to the circa $100 level it was before the fall.
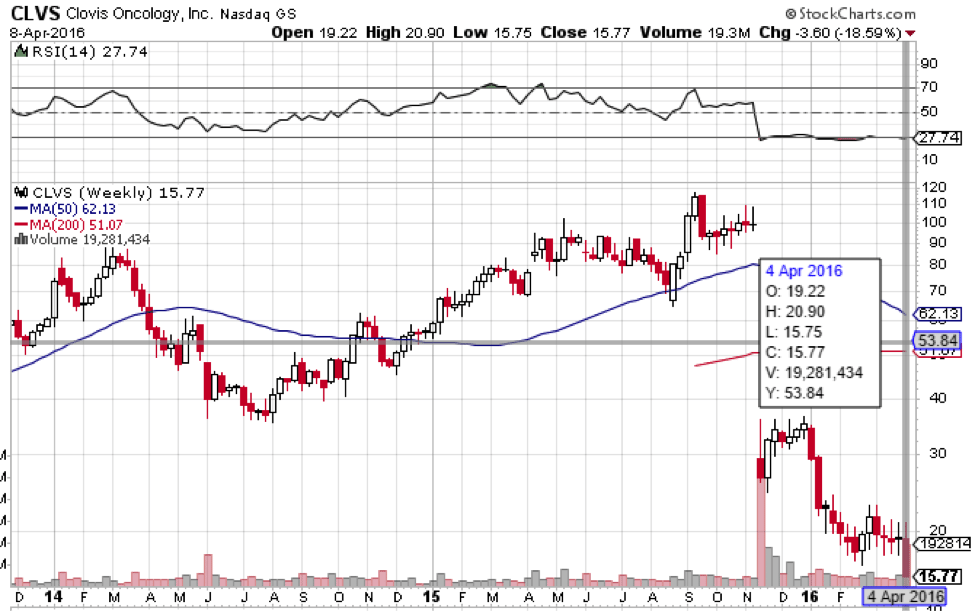
There’s still downside, however. Clovis is heavily invested in Rociletinib, and pretty much its entire valuation is based on the drug’s potential. If the AP recommends non-approval, the floor will fall out from underneath the company’s market capitalization ahead of the FDA’s final decision.
At time of writing, Clovis shares trade for $19.17.
04/15/2016
Chiasma, Inc. (NASDAQ:CHMA)
![]()
FDA decision on Mycapssa for treatment of acromegaly
On April 15, 2016, the FDA will report its final decision on Chiasma Inc’s (NASDAQ:CHMA) Mycappsa. The drug is an acromegaly target. Acromegaly is a condition that comes about as the result of excessive growth hormone production, and leads to a range of unpleasant symptoms including face enlargement, erectile dysfunction, fatigue and – eventually – cardiac and respiratory issues that can lead to premature death.
Mycappsa is a reformulation of the current standard of care, which Chiasma hopes will take over a large percentage of the acromegaly market based on its administration technique. Current SOC is a drug called Sandostatin, and it requires intravenous administration by a physician. It’s painful, overly frequent and – just as with any IV admin treatment – has a host of dangers associated with it. Mycappsa is the same active ingredient, but wrapped in a hydrophilic capsule, which Chiasma designed and has trademarked as Transient Permeability Enhancer (TPE). Because of the protective nature of the TPE, patients can take Mycappsa in pill form, and the active ingredient won’t get broken down and absorbed until it reaches the blood stream, as it does when administered without TPE.
Data showed efficacy to be at least in line with current SOC, and in some cases significantly improved, and safety and tolerability shouldn’t be an issue as the drug is rooted in an already approved compound.
On the surface, there looks to be no real reason why the FDA won’t give Mycappsa the green light come PDUFA on April 15. Of course, there are certain elements of the NDA on which the FDA will base its decision that we are not privy to. There are numerous examples of manufacturing and supply chain issues causing delays – Opko’s (NYSE:OPK) Rayaldee comes to mind here as we covered above – and these might create some unexpected hurdles for the drug.
So what’s the potential upside and downside on approval or turndown? Approval will open up a market worth $1.6 billion annually for Mycappsa. With Chiasma currently valued at a little over $266 million, the potential upside is high double digit, or even triple digit, on an approval. If the FDA issues a complete response, or worse, an outright decline of the drug, expect a 50-75% downside across the session subsequent to the decision’s reporting.
At time of writing, Chiasma shares trade for $10.98.
By: Rafi Farber, Samuel Rae and other Market Exclusive contributors.
Disclosure: At the time of writing this article one of our analysts, Rafi Farber is long shares of Opko.

 Might Be Moving Away From Its Initial Plan With Messenger")
 Is Running Right Now")
: A Misinterpretation Discount?")