
 Chiasma Inc (NASDAQ:CHMA) Sarepta Therapeutics Inc (NASDAQ:SRPT) Collegium Pharmaceutical Inc. (NASDAQ:COLL)")
Market Exclusive Weekly Biotech Report Covering:
- Clovis Oncology Inc (NASDAQ:CLVS)
- Chiasma Inc (NASDAQ:CHMA)
- This Weeks Data
- Sarepta Therapeutics Inc (NASDAQ:SRPT)
- Collegium Pharmaceutical Inc. (NASDAQ:COLL)
The Past Week
Monday April 11, 2016 – Friday April 15, 2016
The week just gone gave us plenty to talk about in the biotech space, with two significant developments – one in oncology and one in a rare genetic growth disorder called acromegaly. Here’s what happened, and how things played out as a result.
Clovis Oncology Inc (NASDAQ:CLVS)
![]()
On April 12, the FDA’s Oncologic Drugs Advisory Committee reported its decision on the company’s lung cancer drug, Rociletinib. The committee advised against approval based on the currently available data, suggesting the agency wait for additional data to surface on the back of an ongoing study called TIGER-3.
The Science
Rociletinib is a third-generation covalent (irreversible) mutant-selective inhibitor of epidermal growth factor receptor (EGFR). The covalent element of this name just refers to the irreversible nature of the bonds the drug forms with proteins as part of its mechanism of action. What is important is the third generation, and the EFGR part.
Non small cell lung cancer (NSCLC) is the most common form of lung cancer, and the current standard of care (SOC) therapies comprise generation one and generation two. Just as does rociletinib, they inhibit the impact of mutant EGFR, and in doing so, inhibit the proliferation of cancerous cells. Sixty percent of NSCLC patients that undergo first and second generation SOC, however, progress, as a result of a gatekeeper mutation – specifically, T790M. Clovis is trying to show that Rociletinib is effective in the inhibition of T790M EGFR induced proliferation.
The Data
The data on which the NDA is based comes from two separate trials, called TIGER-X and TIGER-2. Both investigated the impact that Rociletinib has on patients that have already undergone first and second generation SOC (in this instance, Tarceva or Iressa). The advisory panel voted against approval based on these two trials alone. Instead, it recommended the FDA should wait for the results from an ongoing trial called TIGER-3, which is comparing the effect of Clovis’ candidate to chemotherapy in patients that have both proved positive for the T790M mutation and those that have proved negative for the mutation.
Essentially, the panel is saying that the FDA should see how the drug compares to chemotherapy as well as the current SOC EGFR inhibitors, not just the inhibitors.
The Market
A rival third generation EGFR inhibitor that the FDA approved in November last year – Tagrissa, marketed by AstraZeneca plc (NYSE:AZN) – is priced at $12,750 for a month’s supply. Given that the drug will target the 60% of patients that progress past the first and second generation treatments, this puts the market potential for the drug at circa $3 billion annually. Of course, this would be if Tagrissa didn’t exist, and it does. Clovis will have to compete with AstraZeneca for market share, and unfortunately for the former, it looks as though Tagrissa wins out from an efficacy perspective, meaning Rocelitinib’s actual market potential is far lower than that of Tagrissa.
The Reaction
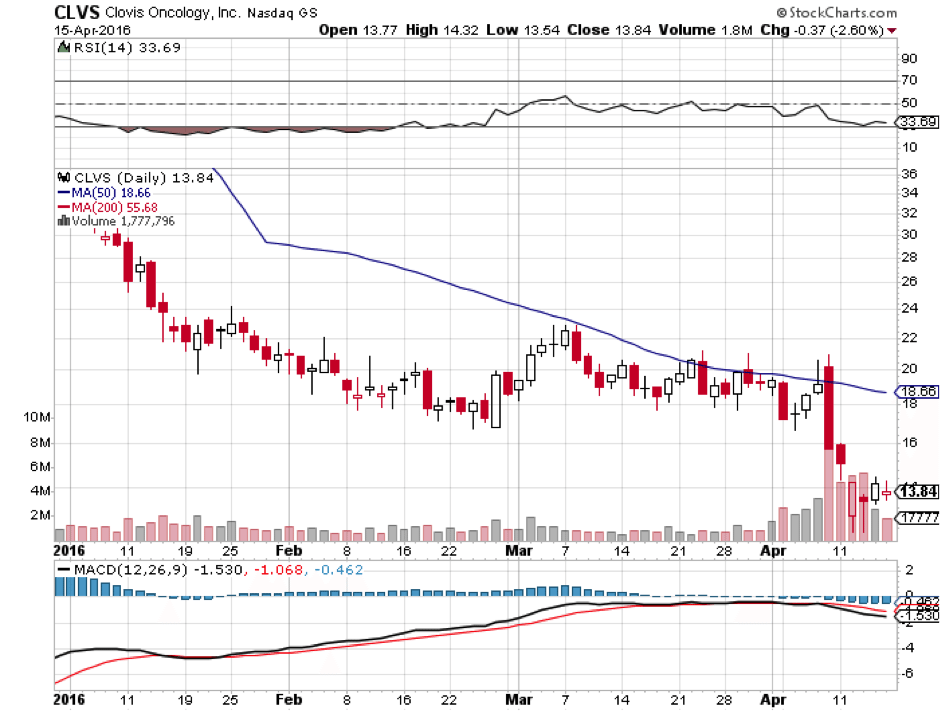
The reaction was not good from a Clovis shareholder perspective. The company lost more than 10% across the session subsequent to the announcement, and remains down as we head into the weekend. A small premarket recovery on Friday has brought the company to trade just ahead of $14 a share.
Finances
If the company fails to get approval when the PDUFA comes around, and the FDA sides with its advisory panel, financing could quickly become an issue for Clovis. The company had $278 million cash on hand at last count, but spends close to that annually on research and development – costs that are likely to ramp up as it progresses its remaining pipeline into late stage development. The company really needed to get generating revenues from its lung cancer candidate as soon as possible, but now it looks like this might be easier said than done.
Valuation and Trading Strategy
The market currently values Clovis at a little over $545 million. Given its cash holdings, this seems like a pretty fair valuation – at the low end of a late stage pipeline company in this sort of position. Having said this, if the FDA turns down Rociletinib, even temporarily while the company completes the ongoing TIGER-3, this $545 million could easily lose a third across a session.
Upside potential on an approval is decent, but given Tagrissa’s likely dominance (at least short to medium term) probably not massive. On approval, a 60-70% upside looks reasonable, though approval chances are probably below 50%. If the FDA declines the drug, expect a 30% to 40% downside on the current valuation.
Chiasma Inc (NASDAQ:CHMA)
![]()
On Friday, April 15, 2016, the FDA reported its decision on Chiasma’s acromegaly candidate, Mycappsa. They issued a complete response letter (CRL), which means no approval yet. The CRL follows data from a phase III trial, submitted as part of an NDA last October.
The Science
Acromegaly is the result of an overactive expression of growth hormone, which leads to excessive growth in various parts of the body, or all parts. Symptoms include large hands, deformed facial features, and a whole host of cardiovascular and respiratory conditions, which eventually lead to death if untreated. Mycappsa takes the current standard of care treatment, or at least the active compound associated with it, and packages it within a capsule. The compound is called octreotide acetate, and it’s not particularly good at reaching its physiological target when administered orally. For that reason, physicians currently use intravenous administration.
In the Mycappsa capsule, however, the drug lasts longer before absorption, and so can be used as an oral administration. The benefits of this sort of administration over an intravenous administration range from being simply more convenient (a patient can administer at home rather than as an outpatient), to cheaper, and of course, safer and less side effect-inducing.
The Data
The data on which the NDA submitted by Chiasma is based investigated the efficacy of Mycappsa across a seven-month period in patients suffering from acromegaly in the US. The endpoint of the trial was normalization of growth hormone (GH) levels, with normalization predefined as < 2.5 ng/mL.
At the end of the trial, the data showed that 65% of patients had GH levels that fell within the predefined normal threshold. At a 13-month measurement of the same sample of patients, the number that had GH levels within the normal threshold was 62%, which accounting for statistical variation, looks pretty consistent with the former data.
The Market
To calculate the market potential for this drug, we can look at the current SOC, the aforementioned intravenous administration, Sandostatin. The drug is now off patent, and so its generic versions are cheaper, but we can assume that if the oral administration Mycappsa is approved, Chiasma can target a price point similar to on-label Sandostatin. At its peak year, Novartis AG (NYSE:NVS), the company that developed and marketed the drug, generated $1.6 billion. We can also assume that an oral administration would quickly penetrate a large portion of the intravenous market, even with the oral administration being more expensive. Given these assumptions, a 50-60% market penetration with a $1.6 billion total market gives a low end $800 million potential revenues, and that’s a conservative estimate.
The Reaction
As yet, reaction to the CRL has not yet been realized. The result came post market close on Friday, and so the true reaction will come as the session opens on Monday. While the company announced that the FDA had issued a CRL, it did not give any further details as to what the CRL says. Chiasma management has scheduled a conference call to discuss the agency’s response before the market opens on Monday morning (8:30 am EDT), and this should offer us some insight into what the FDA wants, and has requested, as part of the process.
Finances
Finances could become a real issue for Chiasma going forward in a couple of scenarios. First, if the FDA has requested further trial data, this would strain the company’s balance sheet (depending on what sort of data, and its required timeframe, a new trial could be very costly), which at last count – December 31, 2015 – only held a little over $41 million cash on hand.
Even if the data required is not particularly drawn out, the delay in being able to generate revenues from Mycappsa means Chiasma will likely need a finance raise near term in order to meet its ongoing operational costs. Dilution, therefore, is a real risk for investors looking to get in at this stage, or that are already exposed to the company.
Valuation and Trading Strategy
The market currently values Chiasma at a little over $246 million, but this will likely prove a very temporary valuation. Why? Because it’s subject to change considerably when Chiasma management discusses the nature of the CRL in its Monday conference call. If the fix is quick, and the company can resubmit at not too high a cost, chances are we will see a little bit of recovery on the Thursday evening, Friday morning decline in the company’s market cap. If it’s a drawn out fix, however, expect a sharp decline, to the tune of 25-35% depending on the specifics.
We can’t really apply an accurate framework for trading ahead of the conference call, other than to say that we will likely see some immediate weakness at market open, and then either a compounding of this weakness if the CRL is severe, or a weak recovery if it’s not difficult a resolution.
The Week Ahead: April 25, 2016 – April 29, 2016
With the FDA decision postponed to July 19th for Progenics Pharmaceuticals, Inc. (NASDAQ:PGNX) and Valeant Pharmaceuticals International, Inc. (NASDAQ:VRX) joint drug Oral Relistor for opioid-induced constipation, this week we will move one more week ahead to April 25th and 26th, covering the FDA panel decision on Sarepta Therapeutics Inc. (NASDAQ:SRPT) Duchenne muscular dystrophy drug eteplirsen, and Collegium Pharmaceutical Inc. (NASDAQ:COLL) tamper-resistant opiate Xtampza.
Sarepta Therapeutics Inc (NASDAQ:SRPT)

On April 25th, and FDA advisory panel will vote on whether it recommends approval for Sarepta’s Duchenne muscular dystrophy (DMD) drug eteplirsen. This will not be the FDA’s final decision as that only comes in May, but a negative vote will almost assuredly doom eteplirsen’s ultimate chances of approval. Eteplirsen has already gone through the FDA wringer and taken a severe beating, but its chances are not yet completely dead.
Back in January, the FDA issued a scathing report on eteplirsen, pretty much admitting that while it was safe, there was no evidence of efficacy. The drug is designed to cause a malfunctioning gene code on exon 51 for the dystrophin protein to be skipped by binding to and blocking that exon from being read, thereby enabling protein synthesis to continue and produce a smaller but functional dystrophin protein. (More on this in the science section.) What follows are key lines from the FDA review in January:
The applicant notes that exon 51 skipping was confirmed by RT-PCR analysis in all patients treated with eteplirsen. PCR is a highly sensitive technique that can detect even a few copies of messenger RNA. Because even a trivial PCR signal is interpreted as “positive,” this biomarker provides little support of efficacy. The RT-PCR finding may, however, provide evidence that eteplirsen leads to at least some degree of exon 51 skipping…
By Western blot, the most accurate quantitative method used by the applicant, mean dystrophin levels after 180 weeks of eteplirsen treatment were reported to be about 0.9% of normal (range <0.25% to 2.5%), with a relative increase of dystrophin of about 3-fold compared to the trace levels typically present in patients with DMD (about 0.3% of normal)…
Eteplirsen-treated patients experienced a sequential worsening of functional abilities and muscle weakness, as demonstrated by changes in rise time from the floor and the evolution of North Star Ambulatory Assessment (NSAA) scores. These two outcome measures are particularly important to the interpretation of the study results. The NSAA has been specifically designed to measure functional ability in ambulatory patients with Duchenne muscular dystrophy,1 and is considered a reliable test that can be used across a range of settings…
The declines in 6 Minute Walk Test over time in eteplirsen-treated patients appear to lie within the range of changes observed in the cohort of patients treated with placebo. Arguably, placebo-treated patients who were blinded to treatment assignment from other controlled trials are more appropriate as matched controls than registry patients, as they may receive special care and attention as trial participants, and may be more highly motivated.
Basically, in January, the FDA panel argued that there is little evidence that eteplirsen provides any clinical benefit to DMD sufferers. The stock plunged as a result, meaning that the shares are currently priced more towards FDA rejection than FDA approval. That doesn’t mean the stock won’t fall further if the FDA panel recommends rejection. It probably will fall further. It does mean though that the upside for Sarepta is much higher if the FDA panel recommends approval, given that the price of the stock is already depressed following the publication of the January report.
The one thing that eteplirsen has going for it is that it is safe, but even that is doubted somewhat due to the size of the efficacy trial. Says the FDA:
No safety signal of significant concern has been identified for eteplirsen, although the clinical safety database for eteplirsen is small, as only 12 patients were exposed for one year or longer, with only 36 patients exposed for 24 weeks or longer (the applicant included safety data from ongoing open-label studies). As a consequence, the one-year database only has adequate power to assess the frequencies of the more common adverse events. Less frequent events, possibly serious, may have been missed because of the small database.
The bottom line is this. The only factor that will sway the FDA panel to recommend approval is the fact that people afflicted with DMD are all going to die, to put it bluntly. These patients are begging the FDA for this drug, even though it is uncertain if it actually works. Approval would essentially mean that the FDA is allowing the drug to be further tested for efficacy in the market itself. Is there a chance of this happening? Yes. To put a number on it, maybe 20% or 30% at most, all depending on the personalities and dispositions of the people on the panel.
When trading becomes a guessing game based on the personal dispositions of a small group of government scientists, it is no longer investing but gambling. So the odds on this bet are about 2:1 in favor of rejection, with a higher payout for approval, making the total odds on this bet about 3:1 or 4:1, roughly.
The Science
Duchenne muscular dystrophy is an extremely rare but devastating genetic disease caused by a mutation in a gene located on the X-chromosome that codes for the dystrophin protein. Because it is located on the X, it typically only affects males, as females have two X’s and at least one of the genes is usually functional. Males only have one X, so if the dystrophin gene is mutated, there are no other copies.
If you imagine a muscle as a chain-link fence, dystrophin acts as the vertical chain link between horizontal muscle fibers, giving them structural integrity and ability to withstand stress through muscle contraction. Without functional dystrophin, muscle fibers rub up against one another and eventually degrade and decay. Patients lose the ability to stand up from the floor, then they lose the ability to walk, and then pulmonary function declines leading to death usually by the mid-20’s.
Eteplirsen is designed to work as follows. DNA codes are composed of code groups called introns and exons. Introns are filler code, and exons are functional. Before DNA codes can be translated into proteins, they are copied by mRNA, and the introns are cut out, or spliced, by molecules aptly called spliceosomes. In DMD, a bad exon displaces the reading frame of the entire remainder of the dystrophin protein. Imagine inserting an extra letter into a sentence and having a computer read the same sentence but with the spacing, or splicing unchanged. For example, we’ll add an extra “a” to the following sentence:
- I am going to the store.
- I aa mgoin gt oth estore .
The extra “a” threw off the reading frame of the whole sentence, with the exon splices being represented by spaces between the words. The spaces are the same after the same amounts of letters, but the words are all gibberish now and the protein doesn’t work. Eteplirsen is designed to do something like this:
- Normal dystrophin reading frame: I am going to the store.
- Dysfunctional dystrophin protein: I aa mgoin gt oth estore .
- Eteplirsen-treated dystrophin: I aam going to the store.
Eteplirsen is supposed to cut out the exon, or word in this case, that contains the initial error, and restore the rest of the reading frame as normal. The result is, or should be, a truncated dystrophin protein that says “I going to the store.” It’s not perfect English, but at least it makes some sense.
How does it actually do this? Eteplirsen is a phosphorodiamidate morpholino oligomer (PMO), which is basically an RNA lookalike. It is designed to mimic the mRNA and bind to it at exon 51 by complementary matching of the bad code. Since the molecule is shaped like an mRNA, it binds to the mRNA at exon 51, preventing that part of the code from being read, causing it to be skipped out, and then the reading frame is restored for the rest of the protein.
The Data
There are two sides to this story. There is the Sarepta side, and the FDA side. The FDA side we already covered. As for the Sarepta side, they believe they found a statistically significant difference in a data point called the Six Minute Walk Test, or 6MWT, in which patients are asked to walk for 6 minutes and see how far they get. Sarepta reports a 151 meter difference for eteplirsen-treated patients. Additionally, the proportion of patients who lost movement was 17% compared to 46% who lost the ability to walk in an external cohort.
Since the 6MWT is only an intermediate endpoint, it needs to be supported by data of dystrophin production in the eteplirsen arm. Sarepta believes it has confirmed evidence of this, and the FDA (sort of) agrees, but Sarepta admits that there is no linear correlation between dystrophin production and clinical outcomes. Still, the fact remains that Sarepta is the only company to demonstrate higher dystrophin levels and a statistically significant difference in the 6MWT.
The problem is, once again, that the trial was not big enough to establish anything definitive, due to the extremely rare nature of this disease. Confirmatory studies are underway, so the FDA may still recommend approval so as not to endanger the ongoing long term open label confirmatory study. Then again, the FDA may recommend rejection and wait for the outcome of the confirmatory study to revisit the topic.
The Market
The market is extremely small for eteplirsen but desperate, which has its advantages and disadvantages. The advantages are that getting to the patients will be relatively easy, and not much marketing will be required. The disadvantage is that the drug will have to be extremely expensive in order to make up the costs of developing it, and the fact that it has not proven efficacy yet could mean that insurance companies will refuse to cover it even if approved.
Exact size is not known with certainty, but here is an extrapolation. About 500 boys per year are diagnosed with DMD. Since they live until about 25 years of age, that means there are about 12,000 boys in the US with DMD at any given time. Eteplirsen is designed to treat DMD originating from exon 51, which is estimated at 13% of the DMD population. That puts the entire eteplirsen market at about 1,560 people, give or take a few hundred.
Sarepta’s accumulated loss since inception is $900 million. They are going to have to make much of that up with a market of 1,560 people. Assuming that all patients take the drug for 20 years, it will have to be priced at about $30,000 annually just to gross that loss over 20 years, which means it will have to be much more expensive if Sarepta wants to actually make money off of it within a reasonable timeframe for its shareholders.
Insurance might not cover that without proof of efficacy, so there’s a big market pickle here even if eteplirsen is approved.
Trading Strategy
Given the odds against approval extrapolated above, and the fact that the shares are priced towards rejection, the best if complicated mathematical strategy here that wouldn’t simply be a guessing game would be an options straddle or a put/call spread weighted proportionally taking into account 3:1 or 4:1 against approval. If eteplirsen is rejected, look for Sarepta shares to halve. If it is approved, shares may triple or more.
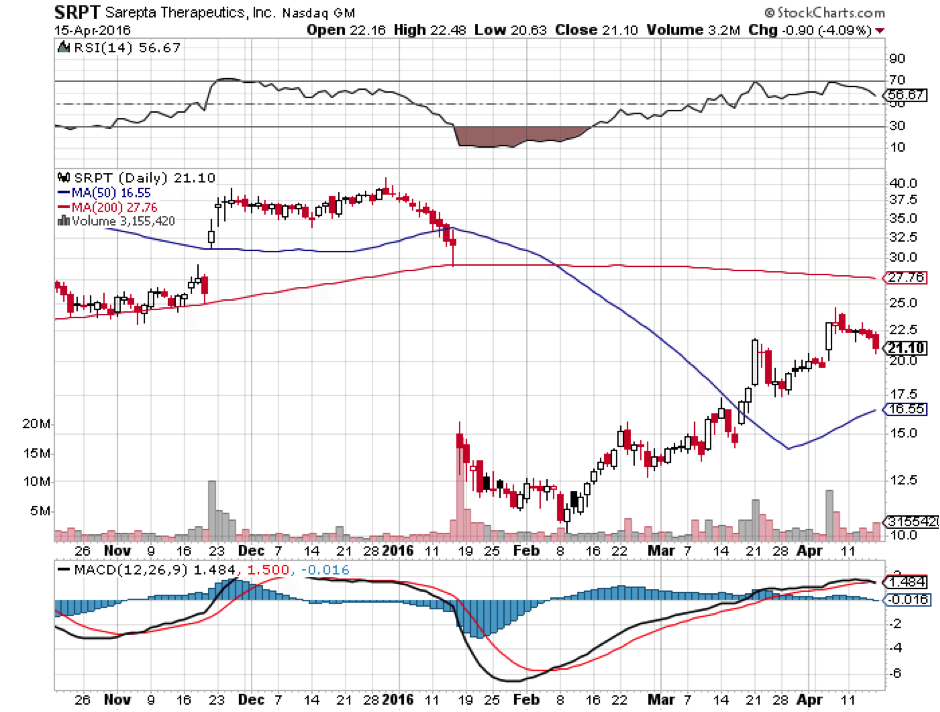
If it is approved and shares do triple though, investors may want to consider taking profits there because there will be market problems with eteplirsen. Sarepta admits this in its last 10-K:
Even with the availability of such [effectiveness] studies, our products may be considered less safe, less effective or less cost-effective than alternative products, and third-party payers may not provide coverage and reimbursement for our product candidates, in whole or in part.
If rejected, the value in Sarepta shares will be based on its continuing phase 3 study for eteplirsen designed to demonstrate long term efficacy.
Collegium Pharmaceutical Inc. (NASDAQ:COLL)

On April 26th, the FDA will issue its final decision on Xtampza, Collegium flagship extended release abuse-resistant opioid candidate for round-the-clock pain treatment. Approval looks very likely if not assured, given that an FDA advisory panel has already voted unanimously to approve the drug back in September, and the FDA granted Xtampza conditional approval in November, with the only snag being an ongoing patent lawsuit by private firm Purdue Pharma L.P.
The only potential issue that could become a problem with Xtampza is the fact that that it needs to be taken with food. Xtampza is essentially tiny extended release pills of oxycodone encapsulated within a bigger pill. Digestion of food helps dissolve the little pills within the bigger pill, so taking it without food tends to diminish the pain relief. This is probably just a minor issue though and will be resolved with labeling.
The Science
Collegium is pretty mum about how Xtampza’s abuse deterrence works, but the general idea is that oxycodone is dispersed evenly in tiny waxy spherical beads. The beads are fatty acid-based, which means they are digested in the small intestine with the digestion of other fats, after which the drug is slowly absorbed. These spherical beads are nearly impossible to crush, dissolve, or chew, so dose dumping, while not 100% impossible, would be expensive to actually do successfully. The point at which dose dumping would be as expensive as buying illegal drugs is the point at which an opiate is effectively unable to be tampered with.
Xtampza can be taken either whole or sprinkled in food or in a feed tube without harming the extended release component.
The Data
The data with Xtampza are pretty straightforward. The drug showed similar efficacy in pain reduction to other opioids with a similar side effect profile. This is not surprising given that it is the same drug, just encapsulated in a fatty acid coat that is eventually digested away. The latest data was published in the journal Pain, focusing on 26 subjects over 65 years of age with chronic lower back pain. 18 of 26 completed the study, and all who completed it showed a clinically important pain reduction from Screening Baseline. All 18 reported improvement, and 14 of the 18 showed a greater than 50% improvement in their chronic lower back pain.
Since approval is a shoe-in for April 26, let’s move on to the market.
The Market
An external marketing study for Xtampza done in 2013 predicted peak revenues for Xtampza at $700 million a year. Oxycodone, the active ingredient in Xtampza, is a blockbuster drug, having sold over $2 billion in 2013. That figure is slowly falling but still blockbuster. Xtampza will be the first abuse-resistant opioid on the market that does not interfere with the active ingredient as part of the abuse-deterrent mechanism.
According to Collegium, 300 pain specialists surveyed by the company, said they would prefer Xtampza over other extended release oxycodone products if access and cost were the same. That makes Xtampza a first line product. Pricing has not been disclosed by the company, but should be similar to OxyContin, if a little more expensive but not significantly so.
Collegium is focusing on the niche geriatrics market initially, as opposed to a general “replace OxyContin as the #1 opioid” approach because that would be biting off more than it can chew. It is therefore focusing on the 1.4 million patients in skilled nursing homes, 35% of whom have chronic pain. OxyContin sees 50,000 prescriptions filled per month in long term care, or $300M a year despite a warning on its package for patients who can’t swallow. Pricing Xtampza equally or similarly to OxyContin could theoretically mean taking most of this $300M a year market in geriatrics within a few years, since Xtampza can be taken with feeding tubes and by patients who cannot swallow, while OxyContin cannot.
In addition to that $300 million, $140 million of OxyContin sells every year in a hospital setting, and some of that market can be taken as well.
The main risk with Xtampza will be Collegium’s ability to sell the drug. They have already expanded their marketing staff to 260 from 46 in 2015, anticipating a product launch this month. The average experience of this sales staff if 6 years in the pain space.
There is also the issue of the Purdue lawsuit, but that does not seem to be a major issue at this point, as one was already dismissed, and the other two lawsuits cannot legally cause another stay in approval.
Finances
Collegium has close to $100 million and believes it is sufficiently capitalized until 2018 when it hopes to launch its second abuse-deterrent morphine candidate based on the same technology.
Valuation and Trading Strategy
The good news is that Collegium’s valuation took a hit when FDA review was postponed back in October, and has not moved significantly since. This would indicate that approval is not yet fully priced in, even though it is nearly assured. That gives good potential upside with less risk than average. The short term risk with Collegium is a quick pullback upon approval due to a combination of profit-taking and the exercise of 1.45 million warrants at an average price of $10.37 as of December 31st, 2015. Those warrants are likely to be exercised, diluting the float by over 20% when approval is finally given. Shares are currently trading at $18.54 as of market open, Monday April 18th.
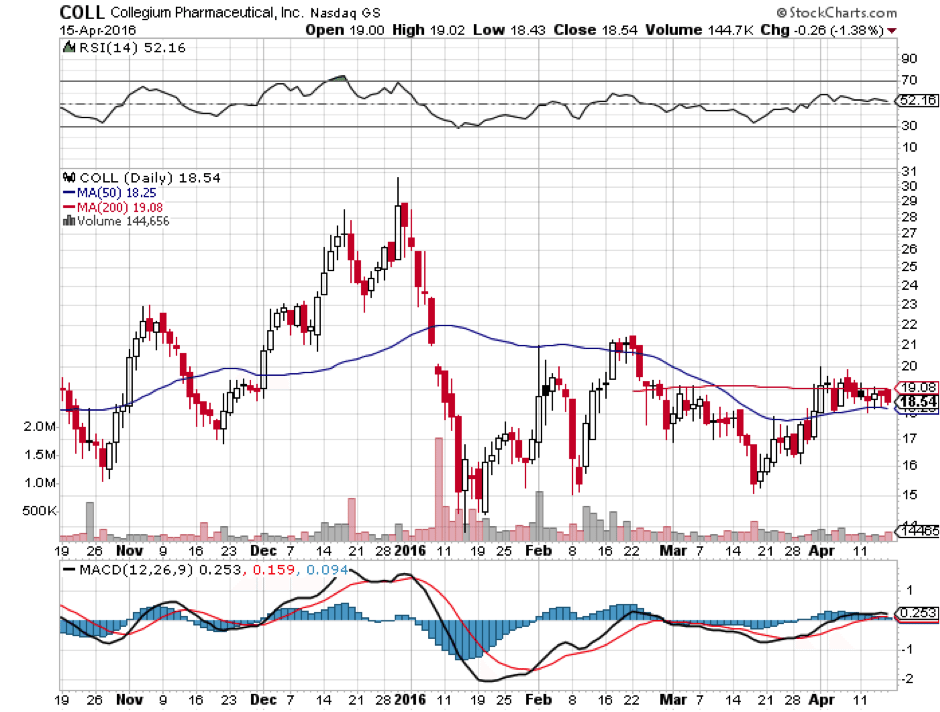 What could very well end up happening is a move at or near January highs of $30.58, about 35% higher than where we are now. This could be followed by a quick selloff though not back down to current levels, due to a combination of dilution and an as-yet unknown market response. The risky way to trade it would be to buy now, and sell into market close post approval, but that is not recommended because good timing in these cases is nearly impossible. The more conservative strategy is to buy a small position now, hold, and add to it on down days until you reach your desired total position.
What could very well end up happening is a move at or near January highs of $30.58, about 35% higher than where we are now. This could be followed by a quick selloff though not back down to current levels, due to a combination of dilution and an as-yet unknown market response. The risky way to trade it would be to buy now, and sell into market close post approval, but that is not recommended because good timing in these cases is nearly impossible. The more conservative strategy is to buy a small position now, hold, and add to it on down days until you reach your desired total position.
If market research is correct and Xtampza can reach a $700 million annual market, we will know first that its technology works, and second that the market accepts it, which will vastly increase the company’s value given its other abuse-deterrent opioids in its pipeline. With a current market cap of $430 million, we could ultimately see a tripling of that by 2018.
Written by: Rafi Farber, Samuel Rae and other Market Exclusive contributors.

 Intercept Pharmaceuticals (NASDAQ:ICPT) Gilead Sciences, Inc (NASDAQ:GILD) Clovis Oncology, Inc. (NASDAQ:CLVS) Chiasma, Inc. (NASDAQ:CHMA)")
 Might Be Moving Away From Its Initial Plan With Messenger")
 Is Running Right Now")