
 Solithromycin Yes Vote Isn’t The Whole Story")
On November 4, 2016, an advisory panel met at the request of the US Food and Drug Administration (FDA) to discuss an antibiotic called solithromycin, currently under development by Cempra Inc (NASDAQ:CEMP). The drug has had a pretty rough road to commercialization, and isn’t quite there yet, but the meeting closed out having voted in favor of approval, and markets interpreted the vote as an indication of the drug picking up an FDA green light come PDUFA.
As it turns out, the vote is not even half the story here.
The real story, and a more accurate investment thesis, is rooted in what happened behind the vote – how responders interpreted the various inputs, and why this led to an, albeit narrow, recommendation.
Here’s what happened.
Before we get into the meeting itself, let’s take a look at the drug and the target indication.
As mentioned, it’s called solithromycin, and it’s targeting a condition called community acquired bacterial pneumonia (CABP). The drug is a macrolide, a type of gram positive therapy that physicians prescribe to treat respiratory tract and soft tissue infections, both of which are associated with CABP. There is a real problem with resistance in the antibacterial space, as is well known, and that extends to pneumococci.
A pneumococcus is just another word for a bacterium that causes pneumonia. Pathogenic pneumococci are the harmful type as most bacteria is harmless, and more than 40% of pneumococci are resistant to the currently available antibiotics.
Cempra is hoping to fill this 60% differential with solithromycin.
It’s not that straightforward, however, as we will discuss shortly. There are some serious concerns surrounding the drug’s relationship with the liver, and the side effects that this can create in certain patients.
This one’s a bit of a strange one. Basically, there are a few things that are in play, but only one that really matters. That the drug is effective in treating patients with CABP, there’s no question. Again, we’ll look at the data that supports this statement in a little more detail shortly, but for now, take our word that this is the case. It’s safety that is the major concern, despite safety trials seemingly identifying no major issues outside of current SOC.
So why is safety a concern if there are no major issues from the current data? It goes back to 2004, and another drug of this type called Telithromycin. Sanofi SA (ADR) (NYSE:SNY) developed Telithromycin, targeting the same indication that Cempra is targeting here, CABP, and the FDA approved it in 2004. In trials, the drug had a great efficacy profile, as measured through clinical response, and what looked to be a pretty clean safety profile. There were some concerns about liver toxicity, but none so serious as to prevent the agency’s green lighting of the drug. Shortly after (within twelve months) patients that took Telithromycin started to develop some serious liver toxicity issues, and 22 died across the period.
It’s since been linked with many many more liver issues, and is currently discontinued in the US. There are a few international administrations, but these are very strictly controlled. Allegations of trial misconduct and the covering up of certain data have since come to light, but we’re not here to discuss that. What’s important is this: the FDA approved a drug of the same class as Solithromycin, and in the same indication, based on what looked like solid data. Shortly after the approval, the drug was directly responsible for the death of numerous patients. The FDA is blamed. The agency took a big hit to its reputation.
The FDA doesn’t want this to happen again, and it’s going to be extra cautious with Cempra’s candidate going forward. Call it cynical, but the FDA cares more about its own reputation than the lives of patients who would otherwise freely choose this drug to save their lives, despite the risks.
There’s another side to the story, however, and it’s this side that Cempra is relying on to make its case. Antibiotics are a finite resource. Bacteria are quickly developing resistance to many of the most widely used, and we aren’t creating them fast enough to offset this resistance. We need new drugs to treat things like CABP, and if the FDA turns potential candidates down based on the fact that they may cause issues that are only identifiable post-marketing, then we may lose the wider war against resistance. We could say a lot more about bacterial resistance, but chances are that readers are already familiar with the issue and its implications.
Here’s what the CDC has to say about it.
Things like TB, and pneumonia, are becoming untreatable, and we need drugs like Solithromycin to hit the market so we can gain control.
These are all things that the panel that convened on November 4 had to take in to consideration. We’ll get to the specific questions shortly, but essentially, the panel had to ask itself the following:
With the potential for liver toxicity deaths very real, can we recommend approval of this drug, or should we turn it down, even though there are wider issues at stake here?
During the meeting, a few different parties weighed in on the discussion, but the two main presentations are what we deem the most relevant as far as preempting the FDA’s decision is concerned. These presentations came from Cempra, and the FDA, respectively.
The FDA kicked things off with a discussion on efficacy, but we won’t get too deep in to that here, because we want to get to the meat of the issue – the potential for liver toxicity. This discussion wouldn’t be complete without an overview, however, so here goes.
Basically, the drug worked.
Solithromycin Efficacy Data
Cempra put it to the test as part of two phase III trials, one based on an oral administration called the 300 study, and another based on IV to oral administration called the 301 study. Both trials were randomized, active controlled, double blind, non-inferiority trials comparing Solithromycin versus Moxifloxacin. Moxifloxacin is a current widely used drug in this indication, and in order to meet the non-inferiority nature of the trial, Solithromycin had to show that it is no worse as defined by a primary endpoint that looked at responder rates than SOC.
Patients were required to be CABP diagnosed with signs, symptoms, and radiographic evidence, and be more than 18 years of age. Primary exclusion criteria included renal failure, severe hepatic impairment, myasthenia gravis, previous hypersensitivity to macrolides, QTprolongation or QT-prolonging drugs. As the exclusion criteria illustrate, the company and the FDA were actively trying to avoid any liver issues going in to the trial, so we can say with some certainty that if the drug does eventually pick up an approval, it will be black boxed to the hilt when it comes to liver concerns.
The primary endpoint was early clinical response (ECR) at a 72-hour visit with some scope on this timeframe, to the tune of -12/+36 hours on either side of the window. ECR was defined by improvement from baseline on ≥2 of the 4 symptoms of cough, dyspnea (difficulty breathing), chest pain, and sputum production, as well as no worsening from baseline on any of the 4 symptoms at ECR visit as measured on a sliding scale at diagnosis. Of course, survival also played in to the endpoint, with survival through the late follow-up visit on Day 28-35 factoring into the primary.
Cempra placed a 10% margin on the trial, meaning that in order to achieve non-inferiority, Solithromycin had to initiate a response in the same number of patients as did moxifloxacin, but with a 10% margin. In other words, if moxifloxacin generates a response in 90% of patients, solithromycin would be required to generate a response in a minimum of 80% of patients to be considered non-inferior.
The image below shows the results of the oral study (300).
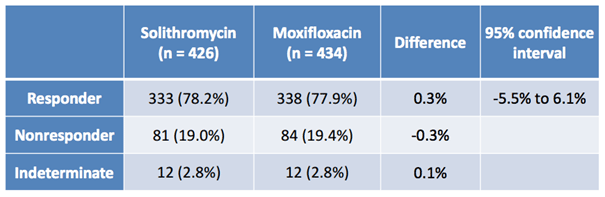
As illustrated, across two arms of circa 430 patients, oral solithromycin generated a slightly better response rate than Moxifloxacin, and the nonresponse rates were both low, and almost identical. From a bull perspective, that’s great news.
The second image, below, shows the results of the IV to oral study (301).
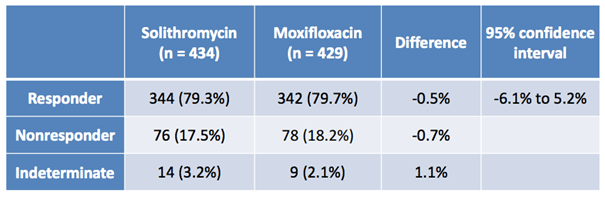
Again, just as with the oral formulation, the data from the IV to oral study shows that the drug performed as well, if not a little better, than Moxifloxacin. Statistical significance is no problem, and indeterminates on both studies aren’t high enough across either arm (active or control) to be relevant to the FDA’s final decision.
The FDA drew the following conclusion in its presentation to the meeting’s attendees, relating to efficacy of the drug when compared to Moxifloxacin:
“The Phase 3 Studies 300 and 301 provided evidence that solithromycin is effective for the treatment of CABP
– Study designs were appropriate for assessing non-inferiority
– Overall efficacy results were similar to moxifloxacin”
So, if we stop there, everyone’s happy and Cempra goes home with what is essentially a guaranteed approval.
Of course, that’s not what happened.
Efficacy established, the FDA went on to present on clinical safety, and it’s this presentation that everybody was there to see. The presentation kicked off with what was essentially a generic discussion of the issues that relate to this class of drugs (myasthenia gravis (MG) exacerbation, tachycardia, visual disorders, syncope) but these aren’t overly relevant to the decision. They can all be avoided with labeling (indeed, the Phase IIIs were set up to exclude patients that might suffer from arguably the most serious of these generic AEs, MG exacerbation) and so won’t really factor in to the panel’s view on approvability, and, ultimately, solithromycin’s chances of commercial approval.
FDA Presentation on Liver Toxicity
Then the agency came to hepatotoxicity, and this is where the attendees really started to sit up and listen. You can witness this yourself on the recording of the meeting. It’s this element of the FDA’s presentation that is going to offer the most insight into what it’s thinking about the approvability of solithromycin.
So, what did we learn?
The agency kicked things off with a discussion of Drug-Induced Liver Injury (DILI). Specifically, are there any identifiable elements of the studies performed to date that might signal DILI, or at least the chances of patients developing DILI within a similar timeframe as telithromycin.
In order to make this assessment, the agency highlighted a number of markers relevant to the signaling. The three markers discussed were ALT elevation, AST elevation and, by proxy, bilirubin elevation. As a reference note, both ALT and AST elevation impair the liver’s ability to clear bilirubin.
So, basically, the question is this: Are there any data points that demonstrate AST or ALT elevation; either elevated levels of both as measured directly, or as inferred by increased bilirubin levels?
The answer is yes, but even with these markers, it’s tough to turn this data into anything that can imply the chances of a drug creating liver issues longer term. We just don’t know enough about the processes that underlie this phenomenon. There is a standardized function, however, that can be used as a sort of best guess system. The function is called Hy’s Law, and the FDA featured its interpretation of Hy’s Law when applied to the solithromycin data, to underpin its safety analysis.
The law looks like this:
ALT/AST elevation ≥3x ULN (upper limit of normal) + total bilirubin (TBL) elevation >2x ULN, without evidence of cholestasis or Any other cause of hepatic injury. This, on the background of higher incidence of hepatocellular injury caused by the drug (AST/ALT ≥3x ULN) compared with the control drug. Such a drug is likely to cause severe DILI (resulting in liver failure or death) at a rate roughly 1/10th the rate of Hy’s Law cases.
Against this framework, no cases of Hy’s Law were identified for solithromycin, but there’s a very important addendum to this conclusion. Using the “Rule of 3’s” in the limited solithromycin safety database, the risk of serious DILI can only be capped at roughly 1:333. The likelihood of severe DILI is known to be much less than that; thus, this database is not large enough to accurately evaluate this risk.
To reword this in a simpler way, normally the application of Hy’s law would infer the chances of DILI. In this instance, however, the safety data base is not large enough to effectively apply the law, or rather, it’s large enough to apply it, but the results are questionable because of the small sample size.
This limitation necessitated the investigation of some of the signals themselves (the above mentioned ALT, AST and Bilirubin), and across these signals, the data did throw up some red flags. Most importantly, and in our opinion probably the most relevant bit of information to come out of the whole development pathway regarding safety is that the AST signal for hepatotoxicity seen with solithromycin across its two phase III trials is greater than what was seen with telithromycin in its own respective phase IIIs over 10 years ago. In other words, the red flag that the FDA missed in its initial approval of telithromycin is present here, and to a higher degree.
Now, in the interest of balance, the chemical composition of solithromycin is slightly different from that of Telithromycin. Cempra uses this point to suggest that a comparison with telithromycin is moot, and that we can’t infer the chances of solithromycin causing DILI in patients using historic telithromycin data. In truth, nobody really knows the answer to this question with any degree of certainty.
To some extent though, the company has a point. The chemical composition is slightly different, and this slight difference does impact the drug’s interaction with the liver. However, the AST signal is there, and in a way, it’s almost worse considering the composition is different. Why? Because it adds some uncertainty to the picture. Specifically, the disparity suggests the potential that solithromycin may trigger additional pathways associated with DILI, pathways not triggered by telithromycin.
Uncertainty is the issue here, and an altered form of hepatotoxicity isn’t doing anything to help alleviate the uncertainty.
So, let’s get to the meat of this discussion. What did the panel say, and what does this mean for the chances of approval?
The Questions and the Vote
Below are the questions posed to each member of the panel, once they had seen and heard what we just discussed:
1. VOTE: Has the Applicant provided substantial evidence of the efficacy of solithromycin for
the treatment of community acquired bacterial pneumonia (CABP)?
a. If yes, please provide recommendations for labeling.
b. If no, please discuss additional studies/analyses that are needed.
2. VOTE: Has the risk of hepatotoxicity with solithromycin been adequately characterized?
a. If yes, please provide any recommendations for labeling.
b. If no, please discuss additional studies that are needed to further characterize the
risk.
3. VOTE: Do the efficacy results of solithromycin for the treatment of CABP, outweigh the
risks including hepatotoxicity?
a. If yes, please provide any recommendations for labeling.
b. If no, please discuss additional studies/analyses that are needed.
For vote 1, the panel voted 13-0. This was expected, and we don’t really need to go into this element of the vote in any more detail. The drug works as measured against its primary endpoint.
For vote 2, the panel voted 12-1 against. To put this another way, the vast majority of the panel believe that the data set isn’t large enough in scope or detail to say with any certainty whether solithromycin is a DILI risk, and more specifically, for whom it might pose this threat.
This wasn’t an unexpected outcome either, and the real surprise came in the fact that one member believes the risk profile is adequately addressed by the data set. Upon further discussion, this member believed labeling could be sufficient to mitigate the risk. Of note, another member suggested that a closely adhered-to follow up might be sufficient to alter their opinion – that is, check in on the patient once or twice a day against the key markers and stop dosing immediately if elevated AST is identified. The practical restrictions make this tough, however.
Vote 3 was the big one, and the panel voted 7-6 in favor of the benefits outweighing the risk profile. This is where Market Exclusive differentiates itself from the wider financial news media. Cempra gained on the back of the drug gaining a slight edge in the panel review, on what is essentially the headline question. For us, however, this doesn’t translate to the company gaining an edge when it comes to the FDA’s final decision. Post vote discussions centered on follow-up trials, as opposed to labeling. Although there were some discussions of labeling, none seemed to completely address the problem at hand.
Now, Cempra could conduct post approval studies, and this in our eyes is the best that the company can really hope for come decision day. However, until these studies are completed, the FDA is going to severely, and we mean severely, limit the market for the drug with some harsh labeling. So much so, that the drug is probably not going to be able to generate any degree of substantial revenues for the company for, let’s say, at least twelve months post approval.
Additionally, and perhaps more importantly, the FDA is not going to want a repeat of the telithromycin situation, and while bacterial resistance is an issue, the bottom line is that current SOCs in the space are still effective right now. In other words, it’s no big deal for the FDA to put off approving the drug for a year or so, while Cempra reinforces its safety data; not in the grand scheme of things.
CABP drugs aren’t going to stop working entirely in 2017, so the FDA has a window of time to collect the data it needs before making a final decision. Obviously, the sooner the better, and not just for shareholders, but the agency doesn’t rush in to a decision, and especially not one on which it has already tarnished its own reputation in the recent past.
Conclusion – Solithromycin Will Likely be Turned Down Despite Positive Vote
So, here’s our take: we think the FDA is going to turn Solithromycin down, demanding a more robust base of safety and tolerability data from Cempra before it gives the drug a green light. This is, of course, bad news for Cempra, but there just doesn’t seem to be any solid argument for approval while there is a chance of disaster post-marketing, when this can be potentially avoided by asking Cempra to wait a few quarters and resubmit. If the current SOC was no longer effective and solithromycin was the only option, then yes, it would probably be approved. But this is not the case, so the FDA will wait and ask for more safety data.

 Intercept Pharmaceuticals (NASDAQ:ICPT) Gilead Sciences, Inc (NASDAQ:GILD) Clovis Oncology, Inc. (NASDAQ:CLVS) Chiasma, Inc. (NASDAQ:CHMA)")
 Might Be Moving Away From Its Initial Plan With Messenger")
 Is Running Right Now")