
 (NYSE:SNY) Delay Highlights Some Red Flags With IGlarLixi")
We just learnt that the FDA has pushed back its decision day on Sanofi SA (ADR) (NYSE:SNY)’s insulin combo candidate IGlarLixi. Markets haven’t moved too much on the news (at least, not yet), but there is a general air of surprise surrounding the delay. Sanofi was pushing to get the product approved ahead of Novo Nordisk A/S (ADR) (NYSE:NVO) competing product, IDegLira, and spent a $245 million priority review voucher ahead of submission to that aim. Now it looks as though there’s going to be practically no difference between the two products’ review dates.
This isn’t the biggest worry for Sanofi, however. The issues that have led to the delay may, in turn, lead to the FDA declining the drug come PDUFA. Here’s why.
The extension is rooted in the device used to administer the combination of insulin glargin and the GLP-1 receptor agonist Lixisenatide. It’s a pen type injectable, with a dial based dosing system. There are two pens available, each of which offer a different min-max dose range.
Here’s what they look like:
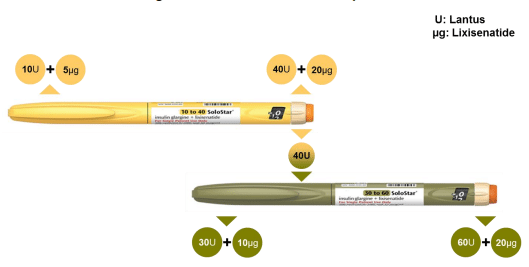
They are designed to deliver a dose based on a patient’s selecting of an insulin dose (as measured in units). The patient uses the scroll mechanism to select their insulin unit number and the pen automatically selects the corresponding dose of Lixisenatide. The yellow pen is a lower dose range (10U-40U) and is targeted at insulin naïve patients, and the green pen is higher dose range (30U to 60U) and targeting those who require higher daily dosing. That all seems relatively simple, right? Well, yes, until we look at the proposed labeling on the scroll system:
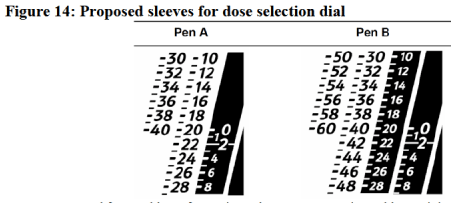
The company has gone for a seemingly strange black/white indication system, designed to show recommended dosing. Doses within the recommended dose range are black writing on white background. Doses below the recommended dose are white writing on black background. The point of this convoluted approach is to discourage dosing errors, but to us, it looks like there is considerable room for human error. The advisory panel agrees:
“It should also be noted that the numbers that are darkened on the pen dial are meant to represent doses that are not established as safe and effective and that should not be used. It is unclear whether this device design element will have the effect it is intended to have (i.e., preventing dosing errors) in the care setting.”
In an attempt to try and overcome this potential hurdle in its NDA, Sanofi submitted data from a human factor trial (HFC) alongside the application. The trial put the pen in front of 60 people (a range of nurses, patients and pharmacologists) and asked them to select the correct dose based on certain individual patient profiles. Here are the results:
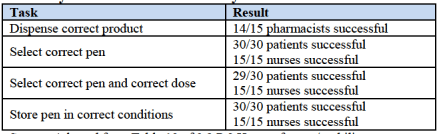
At a glance, these don’t look too bad. However, the advisory panel cited concerns surrounding the study relating to how much time participants had to educate themselves as to usage, and suggested that this might not be representative of a real life situation. Further, that a patient out of thirty mis-dosed, and a pharmacist out of fifteen dispensed the wrong pen, is a concern. At the levels Sanofi is hoping to sell this product, these numbers suggest thousands of individuals mis-dosing.
Compare the Sanofi pen to the Novo pen, and the argument for a total redesign strengthens. The image below is the proposed Novo version, taken from the advisory panel review briefing:
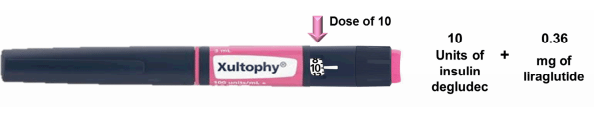
One pen for all doses, and an automatic dose of the GLP-1 agonist. Far simpler, and the review panel recognized this fact, with all members voting unanimously in favor of approval. In contrast, the Sanofi review panel came out 12-2 in favor of an approval. Their concerns?
You guessed it:
I was concerned with the results of the human factor study. I do think the evidence otherwise supports that this agent is effective and safe in the vast majority of patients. I think the advantages outweighed the disadvantages, other than the pen.
I voted ‘no’ solely on the pen design…
Committee member Kenneth D. Burman, MD
Having the wording of the proposed pen in the vote was a struggle for me. The pen needs to be redesigned.
Committee member Ellen W. Seely, MD
According to Sanofi, the application is now resubmitted and the FDA has deemed whatever is included in the resubmission as a major alteration. The fact that it is a major alteration automatically initiates a three-month delay. We are guessing that the alteration is the pen design, but as yet, cannot confirm this.
If it isn’t, Sanofi could be in real trouble come decision day. The FDA has two products that are almost identical in efficacy, tolerability and indication, and it may want to approve both to ensure a competitive landscape and patient choice. However, if one is inferior (and right now, Sanofi’s candidate is) then the agency might refuse to take the risk and decline IGlarLixi outright.

 Intercept Pharmaceuticals (NASDAQ:ICPT) Gilead Sciences, Inc (NASDAQ:GILD) Clovis Oncology, Inc. (NASDAQ:CLVS) Chiasma, Inc. (NASDAQ:CHMA)")
 Might Be Moving Away From Its Initial Plan With Messenger")
 Is Running Right Now")